

Here is a question I was asked: I am a biochemist myself and studied virology many years ago. Unlike HIV, HEP C doesn’t seem to reside in reservoirs. So looking at your SVR rates they are impressive but could you maybe give me a high level reason why treatment fails? Apart from reinfection where could the virus hide? I have bought 20 weeks worth of meds and now I am on week 18. Was undetectable at week 3 so by the end of treatment I will have kept it at bay for a further 17 weeks. They should surely be enough to eradicate it?
Here's the answer.
There are multiple reasons treatment fails. They can broadly be divided into
- Viral Vector Factors
- Host Factors
- Drug Factors
- Resistance and Selective Pressure
Viral Vector Factors
The genetic code of the Hepatitis C virus is single-stranded RNA. Unlike DNA, which has two mirror image strands that deliver error checking, HCV RNA is duplicated like this:
- First, the RNA polymerase makes a mirror image (non-sense) strand of RNA from the viral RNA.
- Then it uses this to make another mirror image (sense) strand.
There is no error checking and 50% of the RNA produced is non-sense. This process is inefficient and error-prone. Every time the RNA duplicates there is about a 1:1000-1:10,000 chance the new RNA copy is inaccurate. Mostly this means it does not work properly, but sometimes it confers resistance. The so-called “wild type” is the most common form of the virus simply because it duplicates itself better than any other variants, however, some of the resistant variants are known as “fit mutants” because these RAVs (Resistance Associated Variants) reproduce almost as well as the wild type. These are the problem children.
Within one person there are a whole range of similar, but slightly different, variants of HCV RNA – this is why it is called a quasi-species.
The RNA itself is used to code a number of proteins of which the NS3/4A, NS5A and NS5B proteins form the 3 different targets for our drugs. The drugs work in a key/lock fashion where the drug is the key and the protein is the lock. The drug keys occupy a vital space in the protein which needs to be vacant for the protein to do its job. Small changes to these proteins change the lock so the key does not fit as well. Small changes to the RNA genetic sequence underlie these changes to the proteins.
Most of our drugs are what is known as competitive inhibitors, so they compete with usual substrate (the thing that normally fits the lock) and therefore need to be above a certain concentration to do their job. This concentration is known as the EC50 (Effective Concentration for 50% of the maximal inhibition). Sofosbuvir works on the NS5B RNA Polymerase protein. The reason it is so good is that this protein is the engine room making new viral RNA and Sofosbuvir is a suicide inhibitor – it binds permanently to the active site so can’t then be displaced when the concentration falls.
The different genotypes of HCV represent similar (but different) RNA sequences. Some genotypes of the virus are naturally resistant to some of the drugs. For example, Ledipasvir does not work well on Genotypes 2 and 3 because the NS5A protein target is a poor fit.
Logically you’d expect the longer you’ve had the virus, and the higher the viral load the more duplication cycles have happened and the greater the chance of resistance. There are hints that this is the case, but no solid proof.
What we do know to be true is that to be resistant to 1 drug is not that hard, but to be resistant to 2 (or more) is much harder. This relates to the 1:1000 probability of a mutation. To get one (on a single strand of viral RNA is 1:1000) but to get 2 is 1:1000 x 1:1000 = 1:1,000,000 and to get 3 it is 1: 1,000,000,000.
HIV is another RNA virus where we have seen this. When we only had one drug resistance happened quite quickly, with 2 drugs it took years, and with current 3+ drug combinations, resistance and virological breakthrough is rare.
Finally, HCV is very infectious. We know from monkey experiments that just 10 virus particles (virions) injected gives a 50% infection probability, and 100 virions deliver a near 100% infection probability. To get rid of this beast we need to kill it down to very nearly the last virion. It’s truly remarkable we can do this.
Host Factors
There is a disease called pernicious anaemia. Patients get this because they cannot absorb the small drug-like molecule vitamin B12. Similarly, some patients do not absorb some drugs very well. For the Hepatitis C DAAs, if we look at the blood levels of a group of patients, the patient with the highest levels may be absorbing 2-10x as much as the patient with the lowest levels. Ledipasvir and Velpatasvir are more problematic in this realm than Sofosbuvir and Daclatasvir.
While the drugs do much of the work, our immune systems still play a role in mopping up the stragglers. We know that 25% of people can clear the virus themselves, and obviously they have strong anti-HCV immune systems. We also know some people run low viral loads – they have moderately strong anti-HCV immune systems. People with high viral loads have weak anti-HCV systems but this is not as bad as you might think. Firstly the virus does not “eat” much so just letting it do its thing causes very little harm (less than fighting it all the time but not quite winning) and with our current drugs people with high viral loads enjoy similar cure rates, albeit sometimes requiring slightly longer treatment.
Unlike HIV there are no known “reservoirs” where HCV can hide, although that said there kind of are. By "kind of are" I’m referring to damaged cirrhotic liver and probably HCC. With cirrhosis parts of the liver are replaced by scar tissue. This tissue has very little blood supply meaning that a drug circulating in the blood does not reach very high levels in this tissue due to the distance it has to diffuse. This makes cirrhotic patients harder to cure, unless we transplant them when, despite the immunosuppression, they are easy to cure. We also see patients with HCC as being harder to cure. This probably relates to the background cirrhosis, but could also relate to the fact that cancer cells refuse to obey apoptosis (die now) signals they get from the immune system.
Drug Factors
As mentioned earlier some drug keys are a poor fit for the protein locks of certain genotypes. This can be a real problem when a patient is co-infected with 2 types of HCV – say genotype 1 and genotype 2. The genotype 1 will be dominant so that patient may get their genotype reported as simply genotype 1, but with ledipasvir not working well on genotype 2, if this patient is treated with Harvoni they will clear the GT1, but relapse with GT2.
Different drugs have different potencies. A drug with a high potency has a low EC50, and a drug with a low potency has a high EC50. For the drug to work it needs to remain above the EC50 throughout the day, otherwise there are times when it is below the level required to contain viral replication. Here is a table for GT3.
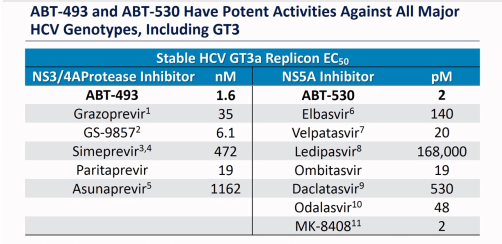
ABT-493 and ABT-530 are the Glecaprevir and Pibrentasvir in Mavyret (Maviret). GS-9857 is the Voxilaprevir in Vosevii and MK-8408 was Ruzasvir (now discontinued).
As mentioned before, some patients do not absorb some drugs as well as other patients so, even if properly dosed they may wander below the critical drug levels.
Some patients do not take their medication every day, and this results in blood levels potentially falling below what is required.
Some medications, supplements and foods interfere with the absorption of the DAAs. Sometimes this increases the blood levels, which, although it leads to more side effects, does not compromise treatment. However, sometimes medications (such as antacids with Harvoni and Epclusa) decrease the blood levels of the drugs and this can lead to treatment failure.
The duration of treatment is vital. The disappearance of the virus follows a pattern of exponential decay meaning (for example) on day 1 we have ½ the virus, day 2 we have ½ of that ½ ie ¼ and on day 3 we have ⅛. Now when we remember we need to go from zillions to < 10 actual viruses we can see that this takes time. Although it saves money we see a lot more GT1 Harvoni failures who took 8 weeks than those who took 12 weeks. The extra weeks are a form of insurance. For almost anyone who achieves SVR at some point we are pouring medication into a body that no longer harbours HCV – we have to overtreat to make sure, and we need to pick a “one size fits all” duration.
Another reason duration is vital relates to the half-life of a viral replicon. This is several weeks. If we don’t treat for long enough a replicon can be suppressed from working, but remove the drugs and it can recover. We need to display no mercy and keep kicking it while it is down until there is no chance it can recover.
Just a quick note on Ribavirin. The log kill on Sofosbuvir is 4.5 meaning it will kill 31,999/32,000. For the NS3/4A and NS5A agents is more like log 3 meaning they will kill 999/1000. Ribavirin is weak in comparison with a log kill of 0.5 meaning it will kill only 2/3 but that may just come in handy mopping up the stragglers. Of note is that Ribavirin is not a DAA – it is a fake letter U in the genetic alphabet so not prone to the same resistance mechanisms as the DAAs.
Resistance and Selective Pressure
We know from HIV that a patient who has a detectable viral load is at risk of a virological breakthrough. Why is that?
When we start treatment, pretty much all the easy to kill virus in the quasi-species is rapidly killed off, however not all of it is. Some of the variants will be partially resistant and the drugs are exerting a selective pressure. Now, despite the fact these RAVs may not be as fit as the wild-type, we have removed the competition. If one of these RAVs “lucks out” and mutates in a way that makes it either more resistant to the current drugs, or more resistant to (the usually missing) 3rd drug on a 3rd target we have a “Houston, we have a problem” kind of a moment.
Wrapping Up
Considering the ask – kill down to the last 10 virions - it’s truly remarkable that Sofosbuvir alone can deliver a ~66% cure rate. We know that adding in an NS5A agent improves that to ~95%. We also see that highly potent NS3/4A+NS5A combinations like Glecaprevir/Pibrentasvir can also kick ass, but here is a lesson from HIV. 3 drugs work better than 2. This is particularly relevant for patients being offered G/P for retreatment. The expected SVR is “only” 90% but if it was me… I’d add in the best NS5B ever invented (Sofosbuvir) to the best NS3/4A and the best NS5B.

{kind=link}